





Abstract
Next generation sequencing (NGS)-based assays continue to redefine the field of genetic testing. Owing to the complexity of the data, bioinformatics has become a necessary component in any laboratory implementing a clinical NGS test.
The computational components of an NGS-based work flow can be conceptualized as primary, secondary, and tertiary analytics. Each of these components addresses a necessary step in the transformation of raw data into clinically actionable knowledge. Understanding the basic concepts of these analysis steps is important in assessing and addressing the informatics needs of a molecular diagnostics laboratory. Equally critical is a familiarity with the regulatory requirements addressing the bioinformatics analyses. These and other topics are covered in this review article.
Bioinformatics has become an important component in clinical laboratories generating, analyzing, maintaining, and interpreting data from molecular genetics testing. Given the rapid adoption of NGS-based clinical testing, service providers must develop informatics work flows that adhere to the rigor of clinical laboratory standards, yet are flexible to changes as the chemistry and software for analyzing sequencing data mature.
Next generation sequencing (NGS)2 is a transformative technology that is redefining the landscape of human molecular genetic testing. It enables unprecedented parallelization of sequencing reactions, facilitating highly multiplexed testing paradigms with relatively rapid turnaround time and decreasing costs (1, 2). A growing number of diagnostic laboratories are embracing NGS and using it to drive new DNA-based test offerings, ranging in size from multigene disease-specific panels (3–6) to entire exomes (7–10) and the rapidly emerging use of complete genome sequencing (11–13). Additionally, applications of NGS for RNA sequencing (14), epigenetic profiling using methylation (15) and chromatin immunoprecipitation sequencing, and microbial and microbiome sequencing (16) offer new avenues for clinical testing. Implicit in clinical adoption of this technology is the need for bioinformatics to process and aid in the interpretation of the massive amount of data generated by the sequencing instruments (17, 18). Bioinformatics is a recently defined discipline that develops and applies advanced computational tools to analyze and interpret high-dimensional biological data. The role of the bioinformatician and bioinformatics work flows are new to clinical sequencing laboratories and require substantial investments in education, personnel, and hardware as well as plasticity in processes and parties involved in the testing.
NGS-based bioinformatics analytics are designed to convert signals to data, data to interpretable information, and information into actionable knowledge. This process can be conceptualized as primary, secondary, and tertiary analyses (19) (Fig. 1). In brief, primary analysis consists of processing raw sequencing instrument signals into nucleotide base and short-read data. Secondary analysis involves the alignment to a reference sequence or de novo assembly of the NGS nucleotide reads and subsequent variant detection, and tertiary bioinformatics analyses provide context to the information generated during an NGS experiment by associating the sample-specific genomic profile with disparate descriptive annotations.
Flow diagram illustrating the major components of a clinical NGS analytical pipeline.
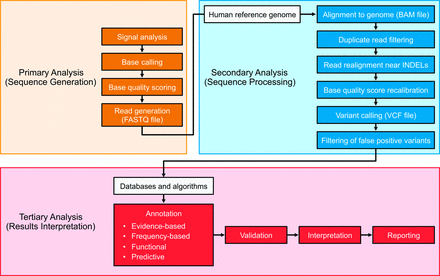
The analysis pipeline is typically considered to comprise the 3 main stages of primary, secondary, and tertiary analysis. Primary analysis generally occurs on-board the sequencing instrument and leads to the generation of genomic sequence in a quality-scored text format. Secondary analysis leads to the detection of variant sequences of various biological categories, and tertiary analysis may include confirmation (or validation) of variants and provides the contextualization of variant information to facilitate interpretation, ideally culminating in clinical decision-making. This general framework varies depending on the precise analytical application, and successful clinical implementation of such work flows requires extensive expertise in bioinformatics and clinical regulatory issues.
The goal of this review is to increase awareness of the different aspects of bioinformatics analysis and their associated regulatory requirements and hurdles. Given the current predominance of DNA-based sequencing in the clinical arena, this review focuses on human DNA-based bioinformatics analysis. It will describe several of the more popular bioinformatics solutions and draw attention to some of the potential pitfalls and challenges that reside in this field. A comprehensive review of all possible bioinformatics solutions is beyond the scope of this report, and readers interested in broader coverage are directed to several extensive bioinformatics reviews (20, 21). In this review we first describe the nature of primary, secondary, and tertiary analyses and then discuss the current regulatory landscape, with emphasis on defined requirements, before providing a forward-looking summary of the current informatics challenges and emerging solutions that will drive more advanced clinical sequencing applications.
Bioinformatics Processes
SEQUENCE GENERATION (PRIMARY ANALYSIS)
Primary analysis is a process that has become highly integrated with the sequencing instruments and associated onboard software. These tools convert the raw signals generated by the sequencing instruments into nucleotide bases with associated quality scores, and ultimately, short nucleotide sequences or “reads.” In some instances, the primary analysis also includes demultiplexing of multiple samples indexed and pooled into a single sequencing run. Primary analysis software is provided by all major sequencing vendor companies and often is installed on the hardware systems supporting the sequencing instruments. However, it can also be offloaded onto high-performance clusters or cloud-based architectures for improved performance or iterative analyses. To date, there has been limited development of independent primary analysis software programs, and as such this topic is not covered in detail. However, if NGS software evolves similarly to microarray analysis software, this could become an area of latent focus as software developers strive to improve the initial signal processing in attempts to improve overall data integrity; therefore, further software developments should be closely monitored.
ALIGNMENT AND VARIANT DETECTION (SECONDARY ANALYSIS)
Secondary analysis consists of a variable collection of methods that operate together to detect genomic aberrations from quality-scored sequence data. Depending on the protocol, this profiling can occur at the level of the genome, exome, or focused gene panels. The class of genomic variation profiled can vary and includes single nucleotide variants, small insertions and deletions, or larger alterations like structural rearrangements and copy number changes (Table 1). Furthermore, genomic variations can be either constitutional (de novo or inherited) or somatic (acquired), affecting only a subset of the body's cells, such as in cancer. Although each of these considerations introduces subtle differences to the analysis protocol, the fundamental processes are highly similar.
Commonly used tools for NGS-based DNA analysis by functional category.a
A selection of commonly utilized NGS analysis tools used in secondary analysis work flows. All tools are freely available, although licensing may be necessary to enable for-profit use.
CNV, copy number variation; SV, structural variant.
A selection of commonly utilized NGS analysis tools used in secondary analysis work flows. All tools are freely available, although licensing may be necessary to enable for-profit use.
CNV, copy number variation; SV, structural variant.
The initial secondary analysis step usually involves the collective alignment of reads to a reference human genome. De novo assembly of a genome is also possible but currently less common in human applications owing to the existence of a high-quality reference genome and the more experimental nature of genome assembly methods (22, 23). Once reads have been aligned to the genome, several refinement steps are often performed (24). These steps routinely include flagging or filtering of duplicate reads likely to be PCR artifacts, and realignment, which leverages a collective view of reads around putative insertion/deletion (indel) sites to minimize erroneous alignment of read ends. Quality scores allocated by the sequencing software will often be recalibrated on the basis of alignment data, before proceeding to the variant calling stage. Variant calling involves the comparison of the sequenced reads to their point of alignment on the human genome to determine areas that differ on the basis of statistical modeling techniques that aim to distinguish genuine genomic variations from errors (25). In general, specialized programs are selected dependent on the class of variant being investigated. Sizes of events—ranging from a single nucleotide base pair to many millions—affect how individual software performs, because a particular algorithm may be tuned to detect only one type and size of event. Oncology applications often differ methodologically in that they involve separate comparisons of normal and tumor samples to the reference genome, and subsequent analysis of the differences between the two enable selection of tumor-specific variation. Variant calling errors are common, as NGS technologies are inherently less accurate than traditional sequencing methods and, therefore, artifacts occur with greater regularity (26). This problem is partially corrected for by increasing sequencing depth (i.e., sequencing each base position multiple times). An example of how multiple reads are aligned to the same genomic region, thereby increasing sequencing depth, is illustrated in Fig. 2. The use of high-depth sequencing is particularly powerful in panel-based approaches in which the query region is small and great depths can be attained. In comparison, exome- and genome-sequencing efforts are complicated by the increased target region size and issues such as variable capture or sequencing efficiency, which collectively introduce regions of insufficient sequence depth and increase validation burden (27). Repetitive genomic regions and pseudogenes introduce alignment ambiguities due to the relatively short read lengths generated by most NGS technologies, and this represents another source of error (28).
Potential effects of multiple transcript isoforms on variant effect prediction. The image shows a screenshot of NGS data within Integrative Genomics Viewer (IGV) [Thorvaldsdottir et al. (76)].
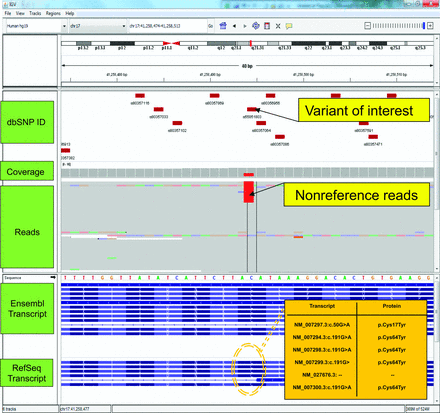
The top panel displays a chromosome ideogram with a red block highlighting the region being displayed in the lower panels. The genomic coordinates beneath the ideogram describe the precise genomic location of the current view. In this case, the view spans a 61-bp window, ∼41 Mb into chromosome 17 at q21.31. Beneath the coordinates, 5 separate data tracks are displayed. The first track contains the positions and IDs of dbSNP entries. Below these are the depth of coverage and read alignment tracks for a single biological sample. Note the colored regions in the read alignment track, which represent nucleotides that differ from the hg19 reference; in this case showing a C>T transition. Exonic structure of transcripts derived from the RefSeq and Ensembl databases are displayed in the lower panels. The HGVS nomenclature for the highlighted variant is c.191G>A (p.Cys64Tyr) or c.50G>A (p.Cys17Tyr), depending on the transcript affected. Such discrepancies have the potential to confuse variant interpretation particularly in instances where data are inappropriately collapsed and summarized as a single amino acid change, potentially omitting predictions of pathogenicity. Note, the HGVS nomenclature is represented as G>A rather than C>T because this gene is oriented on the reverse genomic strand.
Individual sequencing technologies often suffer from platform-specific error profiles (29), which can only be partially anticipated and corrected before generation of a variant call set. Consequently, erroneous variant calls inevitably occur, and thus filtering or confidence-based prioritization of variant calls is a key component of the secondary analysis work flow. Prioritization is often preferred to removal of candidate variants to avoid the incorrect and irreversible filtering of a genuine variant call. The filtering or prioritization process can involve computational or human efforts, including visual inspection of variant alignments, and can be based on empirical cutoffs or more advanced statistical approaches. Criteria used to assess the quality of variant calls varies but examples include the frequency with which a variant allele is observed in a sample, the base quality of the variant alleles as predicted by the sequencing instrument, and the ability of a read containing a variant allele to map uniquely to a single location on the human reference genome. A unique challenge is posed by oncology-based applications in which sample heterogeneity (multiple tumor clones) and purity (normal tissue contamination of a tumor sample) further confound reliable variant calling by altering the expected frequency of observation of variant alleles. Carefully considered and characterized filtering or prioritization cascades therefore must be implemented in any analysis approach to attain acceptable and reproducible levels of sensitivity and specificity.
Implementation of a pipeline that encapsulates each of the analysis steps described is a nontrivial task. An initial obstacle is the necessity for both sufficient computational hardware and staff with the appropriate technical knowledge to operate both the hardware and the requisite software. Genomic analysis pipelines are computationally intensive and implemented solutions must be capable of running on available hardware and doing so in a time frame amenable to clinical turnaround. In addition to these considerations, bioinformatics challenges are numerous and the field is thus a highly dynamic area of research (Table 2) (30). Multiple open source or commercial software solutions invariably exist for any single analysis step, each with their own characteristics, strengths, and weaknesses (19). Often individual software applications are tailored to a particular sequencing platform, sequence length, or sequencing protocol (31, 32). Alternatively, several applications might be suited to identical data types but perform very differently. Sequence aligners are perhaps one of the most numerous software solutions, and for clinical applications, understanding the differing performances (33, 34) and nuances of aligners and subsequent impacts on all remaining sequence analyses is critical. Germline and somatic variant callers are also widely recognized as generating very different results and often each caller will detect its own distinct set of unique, correct calls (31, 35). These variations in performance also affect algorithms designed to detect copy number variations and other larger-scale alterations (36). Additionally, bioinformatics solutions are often highly customizable and their performance is exquisitely sensitive to their correct parameterization. With these facts considered it is therefore unsurprising that alternate pipelines have been shown to disagree to a great extent (37).
Commercially available integrated DNA analysis solutions and relevant functionality.a
| Applications | |||||||
|---|---|---|---|---|---|---|---|
| Read alignment | SNVb detection | INDEL detection | SV detection | CNV detection | Variant annotation | Visualization | |
| CLCBIO Genomic Workbench |  | | | | | | |
| Softgenetics Nextgene | | | | | | | |
| Genomatix | | | | | | | |
| DNAStar Lasergene | | | | | |||
| Avadis NGS | | | | | | | |
| Applications | |||||||
|---|---|---|---|---|---|---|---|
| Read alignment | SNVb detection | INDEL detection | SV detection | CNV detection | Variant annotation | Visualization | |
| CLCBIO Genomic Workbench | | | | | | | |
| Softgenetics Nextgene | | | | | | | |
| Genomatix | | | | | | | |
| DNAStar Lasergene | | | | | |||
| Avadis NGS | | | | | | | |
A selection of commonly implemented commercial integrated analysis solutions. The table focuses on functions relevant to the work flows described within this review and each solution has functionality beyond that listed here.
SNV, single nucleotide variant; SV, structural variant; CNV, copy number variation.
| Applications | |||||||
|---|---|---|---|---|---|---|---|
| Read alignment | SNVb detection | INDEL detection | SV detection | CNV detection | Variant annotation | Visualization | |
| CLCBIO Genomic Workbench | | | | | | | |
| Softgenetics Nextgene | | | | | | | |
| Genomatix | | | | | | | |
| DNAStar Lasergene | | | | | |||
| Avadis NGS | | | | | | | |
| Applications | |||||||
|---|---|---|---|---|---|---|---|
| Read alignment | SNVb detection | INDEL detection | SV detection | CNV detection | Variant annotation | Visualization | |
| CLCBIO Genomic Workbench | | | | | | | |
| Softgenetics Nextgene | | | | | | | |
| Genomatix | | | | | | | |
| DNAStar Lasergene | | | | | |||
| Avadis NGS | | | | | | | |
A selection of commonly implemented commercial integrated analysis solutions. The table focuses on functions relevant to the work flows described within this review and each solution has functionality beyond that listed here.
SNV, single nucleotide variant; SV, structural variant; CNV, copy number variation.
The array of available solutions and lack of established gold standards creates difficulties when considering the appropriate toolset for clinical applications. Each component of a pipeline must be carefully selected and its performance characterized, compared, and validated. This fact is well understood in the field and software is recognized as an independent area of validation in quality guidelines for NGS technologies (38, 39). Commercially available analysis solutions often form core components of clinical work flows, at least partially, owing to the reduced validation burden they impose on a laboratory. Nonetheless, there is often a trade-off in innovation vs stability when considering open-source bioinformatics software or commercially vended solutions. It is possible under some circumstances that no one approach or configuration will be sufficient to achieve acceptable performance for a given application, and in such instances the use of parallel and complementary methods is advisable in many cases to achieve the required level of sensitivity.
ANNOTATION AND VISUALIZATION (TERTIARY ANALYSIS)
Following detection, variants must be annotated to determine their biological significance and enable functional prioritization and downstream interpretation. This characterization is generally achieved using a combination of biological annotation sources including frequency-, structural-, prediction-, or evidence-based data. Each class of annotation has associated benefits and limitations, and when applied in subsequent interpretation can introduce further analytical challenges. Several key resources used to annotate NGS data are described in Table 3.
Commonly used tertiary analysis annotation resources.
| Annotation source | Description | Available via | |
|---|---|---|---|
| Population frequency based | 1000 Genomes Project | Low-coverage whole genome sequencing of 2500 healthy humans | http://www.1000genomes.org |
| NHLBI Cohort | 6500 Sequenced exomes from heart, lung, and blood disorder patients | https://esp.gs.washington.edu/drupal/ | |
| HapMap Project | SNPa-based data set to define haplotypes across 270 ethnically diverse humans | http://hapmap.ncbi.nlm.nih.gov | |
| Structural based | SnpEff | Variant impact on codon and gene structure | http://snpeff.sourceforge.net/SnpEff.html |
| VEP | Variant impact on gene, transcript, protein sequence | http://www.ensembl.org/info/docs/tools/vep/index.html | |
| Prediction based | SIFT | Sequence conservation | http://sift.jcvi.org/ |
| POLYPHEN | Phylogenetic and structural characteristics | http://genetics.bwh.harvard.edu/pph/ | |
| CONDEL | Meta-prediction aggregator | http://omictools.com/sequencing/genome-resequencing/driver-mutations/condel-s654.html | |
| MutPred | Random forest prediction method | http://mutpred.mutdb.org/ | |
| CADD | Meta-prediction and annotation score | http://cadd.gs.washington.edu | |
| VAAST | Phylogenetic and disease-based conservation | http://www.yandell-lab.org/software/vaast.html | |
| MutationTaster | Meta-data type integration | http://www.mutationtaster.org | |
| ANNOVAR | Meta-data, meta-prediction aggregator | http://www.openbioinformatics.org/annovar/ | |
| Evidence based | OMIM | Disease phenotype–gene relationships | http://www.omim.org |
| Leiden Open Variation Database | http://www.lovd.nl/3.0/home | ||
| Human Gene Mutation Database | Human inherited disease gene lesions | http://www.hgmd.org | |
| ClinVar | Clinical human variation to phenotype relationships | http://www.ncbi.nlm.nih.gov/clinvar/ |
| Annotation source | Description | Available via | |
|---|---|---|---|
| Population frequency based | 1000 Genomes Project | Low-coverage whole genome sequencing of 2500 healthy humans | http://www.1000genomes.org |
| NHLBI Cohort | 6500 Sequenced exomes from heart, lung, and blood disorder patients | https://esp.gs.washington.edu/drupal/ | |
| HapMap Project | SNPa-based data set to define haplotypes across 270 ethnically diverse humans | http://hapmap.ncbi.nlm.nih.gov | |
| Structural based | SnpEff | Variant impact on codon and gene structure | http://snpeff.sourceforge.net/SnpEff.html |
| VEP | Variant impact on gene, transcript, protein sequence | http://www.ensembl.org/info/docs/tools/vep/index.html | |
| Prediction based | SIFT | Sequence conservation | http://sift.jcvi.org/ |
| POLYPHEN | Phylogenetic and structural characteristics | http://genetics.bwh.harvard.edu/pph/ | |
| CONDEL | Meta-prediction aggregator | http://omictools.com/sequencing/genome-resequencing/driver-mutations/condel-s654.html | |
| MutPred | Random forest prediction method | http://mutpred.mutdb.org/ | |
| CADD | Meta-prediction and annotation score | http://cadd.gs.washington.edu | |
| VAAST | Phylogenetic and disease-based conservation | http://www.yandell-lab.org/software/vaast.html | |
| MutationTaster | Meta-data type integration | http://www.mutationtaster.org | |
| ANNOVAR | Meta-data, meta-prediction aggregator | http://www.openbioinformatics.org/annovar/ | |
| Evidence based | OMIM | Disease phenotype–gene relationships | http://www.omim.org |
| Leiden Open Variation Database | http://www.lovd.nl/3.0/home | ||
| Human Gene Mutation Database | Human inherited disease gene lesions | http://www.hgmd.org | |
| ClinVar | Clinical human variation to phenotype relationships | http://www.ncbi.nlm.nih.gov/clinvar/ |
SNP, single nucleotide polymorphism.
| Annotation source | Description | Available via | |
|---|---|---|---|
| Population frequency based | 1000 Genomes Project | Low-coverage whole genome sequencing of 2500 healthy humans | http://www.1000genomes.org |
| NHLBI Cohort | 6500 Sequenced exomes from heart, lung, and blood disorder patients | https://esp.gs.washington.edu/drupal/ | |
| HapMap Project | SNPa-based data set to define haplotypes across 270 ethnically diverse humans | http://hapmap.ncbi.nlm.nih.gov | |
| Structural based | SnpEff | Variant impact on codon and gene structure | http://snpeff.sourceforge.net/SnpEff.html |
| VEP | Variant impact on gene, transcript, protein sequence | http://www.ensembl.org/info/docs/tools/vep/index.html | |
| Prediction based | SIFT | Sequence conservation | http://sift.jcvi.org/ |
| POLYPHEN | Phylogenetic and structural characteristics | http://genetics.bwh.harvard.edu/pph/ | |
| CONDEL | Meta-prediction aggregator | http://omictools.com/sequencing/genome-resequencing/driver-mutations/condel-s654.html | |
| MutPred | Random forest prediction method | http://mutpred.mutdb.org/ | |
| CADD | Meta-prediction and annotation score | http://cadd.gs.washington.edu | |
| VAAST | Phylogenetic and disease-based conservation | http://www.yandell-lab.org/software/vaast.html | |
| MutationTaster | Meta-data type integration | http://www.mutationtaster.org | |
| ANNOVAR | Meta-data, meta-prediction aggregator | http://www.openbioinformatics.org/annovar/ | |
| Evidence based | OMIM | Disease phenotype–gene relationships | http://www.omim.org |
| Leiden Open Variation Database | http://www.lovd.nl/3.0/home | ||
| Human Gene Mutation Database | Human inherited disease gene lesions | http://www.hgmd.org | |
| ClinVar | Clinical human variation to phenotype relationships | http://www.ncbi.nlm.nih.gov/clinvar/ |
| Annotation source | Description | Available via | |
|---|---|---|---|
| Population frequency based | 1000 Genomes Project | Low-coverage whole genome sequencing of 2500 healthy humans | http://www.1000genomes.org |
| NHLBI Cohort | 6500 Sequenced exomes from heart, lung, and blood disorder patients | https://esp.gs.washington.edu/drupal/ | |
| HapMap Project | SNPa-based data set to define haplotypes across 270 ethnically diverse humans | http://hapmap.ncbi.nlm.nih.gov | |
| Structural based | SnpEff | Variant impact on codon and gene structure | http://snpeff.sourceforge.net/SnpEff.html |
| VEP | Variant impact on gene, transcript, protein sequence | http://www.ensembl.org/info/docs/tools/vep/index.html | |
| Prediction based | SIFT | Sequence conservation | http://sift.jcvi.org/ |
| POLYPHEN | Phylogenetic and structural characteristics | http://genetics.bwh.harvard.edu/pph/ | |
| CONDEL | Meta-prediction aggregator | http://omictools.com/sequencing/genome-resequencing/driver-mutations/condel-s654.html | |
| MutPred | Random forest prediction method | http://mutpred.mutdb.org/ | |
| CADD | Meta-prediction and annotation score | http://cadd.gs.washington.edu | |
| VAAST | Phylogenetic and disease-based conservation | http://www.yandell-lab.org/software/vaast.html | |
| MutationTaster | Meta-data type integration | http://www.mutationtaster.org | |
| ANNOVAR | Meta-data, meta-prediction aggregator | http://www.openbioinformatics.org/annovar/ | |
| Evidence based | OMIM | Disease phenotype–gene relationships | http://www.omim.org |
| Leiden Open Variation Database | http://www.lovd.nl/3.0/home | ||
| Human Gene Mutation Database | Human inherited disease gene lesions | http://www.hgmd.org | |
| ClinVar | Clinical human variation to phenotype relationships | http://www.ncbi.nlm.nih.gov/clinvar/ |
SNP, single nucleotide polymorphism.
Comparison of an individual's genome to the current human reference sequence will produce many variant calls that essentially represent benign interindividual human variation. Population frequency–based annotations are often a core component of tertiary analysis because variants that are common in the general population are unlikely to have biological relevance in the context of a clinical assay. Frequency thresholds are generally applied to remove benign polymorphisms from variant lists. These thresholds may be set differently depending on the assay. For instance, a suspected fully penetrant autosomal dominant mutation is likely to be absent from population-based cohorts. Conversely, inherited variants predisposing carriers to increased breast cancer risk would be expected to occur at a greater frequency in the population and therefore a less stringent threshold might be employed. Common sources of frequency-based annotations include the 1000 genomes project (40) and the NIH National Heart, Lung, and Blood Institute (NHLBI) cohort (41), as well as laboratory-specific control samples or internal databases. Each data set includes its own biases dependent on the sample and sample characterization included in the resource. For example, the only phenotype information available for the 1000 genomes data set is that individuals were “healthy” at the time of collection. The NHLBI cohort meanwhile comprises both healthy control samples and extreme phenotypes including increased blood pressure and increased risk of myocardial infarction. Therefore, population composition may need to be controlled for in instances in which total frequency in the cohort is not appropriate. Laboratory-specific controls refer to samples from within an institution that have gone through identical primary and secondary analysis pipelines. These controls account for work flow–specific variants that may appear due to differences in bioinformatics algorithms in the assay vs those used in the larger cohort studies (42).
Structural-based annotations assign the effect of a variant on the transcripts and encoded protein(s) based on the resulting amino acid change (43, 44). The effect on the encoded protein sequence is subsequently categorized using clearly defined rules; for example, nonsense mutations are categorized as highly impactful. Tools which assign structural-based variant effects generally also provide annotations including (but are not limited to) approved Human Genome Variation Society (HGVS) nomenclature for a variant, region of a transcript affected, and the likelihood of a variant initiating nonsense-mediated decay. Importantly, many genes produce multiple transcripts that create the possibility of multiple conflicting HGVS-format amino acid changes being associated with a single genomic alteration. This can confuse variant annotation and interpretation, due to differing predictions of effect existing for a single variant. Fig. 2 illustrates this concept using a potentially pathogenic breast cancer 1, early onset (BRCA1)3 allele for hereditary breast and ovarian cancer as an example. Depending on the transcript used, the amino acid change could be correctly reported as either Cys17Tyr or Cys64Tyr. Caution is therefore necessary when interpreting variant effects based on amino acid location without transcript information.
Differences existing between reference genome and transcript sequences pose another challenge to accurate structural-based annotation. As of April 7, 2014, there were 6620 instances of site-specific nucleotide base differences between transcripts in the human Reference Sequence (RefSeq) transcript database and human reference genome version hg19 (GRCh37), affecting 5308 transcripts from 3039 genes. Such instances can cause erroneous variant calls and incorrect downstream interpretation. Clinically relevant genes affected by such discordances include the ABO blood group gene, the oncogene v-akt murine thymoma viral oncogene homolog 1 (AKT1), and the pharmacogenetically relevant genes cytochrome P450, family 2, subfamily C, polypeptide 19 (CYP2C19) and cytochrome P450, family 2, subfamily D, polypeptide 6 (CYP2D6).
Prediction-based annotations use nucleotide and/or amino acid changes integrated with additional contextual data, including evolutionary conservation scores, amino acid substitution matrices, and impact on 3D protein structures to infer the variant's impact on the resulting sequence product. These software systems often use computational learning models (e.g., neural networks, decision trees, Hidden Markov Models) or, more recently, integrated metaanalyses from existing systems to produce the resulting annotations (45–52). The sensitivity and specificity of these tools, however, leaves much to be desired (53, 54). Even empirical or Mendelian prediction models based on a single gene have different levels of accuracy (55), highlighting the difficulty of performing such a task on a genome-wide scale including many genes with unknown function.
Finally, evidence-based annotations are derived from the literature and other historical data. Large-scale projects have been underway for some time to catalog and further curate variants associated with diseases (56, 57). Evidence-based annotations might intuitively be expected to be the most reliable; however, as many as 27% of variants published as pathogenic are incorrectly classified (58). To repair this classification error and to centralize these fragmented databases, the newly initiated ClinVar (59) aims to centralize the collection of clinically actionable (as well as truly benign) germline variants and standardize their reporting according to the recommendations by the American College of Medical Genetics and Genomics (60).
There are increasing numbers of commercial applications designed to facilitate NGS result annotation and data interpretation (Table 4). When variant data are uploaded into the software, annotations are automatically added through remote and/or local databases. Many out-of-the-box solutions are functionally similar, providing an interactive graphical user interface that enables review of results, simple and complex data queries, and graphical display of data using some form of genome browser. These systems are often customizable to enable addition of features and database access. Some applications also allow users to view alignments and variants simultaneously, which aids in technical QC. Annotations commonly include information from the OMIM (Online Mendelian Inheritance in Man) database, dbSNP (the Single Nucleotide Polymorphism Database), and the NHLBI cohorts. Where these commercial applications differ is the inclusion of graphical interfaces for aligned read review, the number of public annotations integrated, and the ability to capture and recall user-specified variant descriptions. A few of the more advanced tools will also allow curation of local results and retrieval of this information for future decision-making, a powerful learning feature that increases the efficiency of data review.
Commercial applications that integrate multiple NGS annotation resources into a user-friendly environment for variant review, visualization, and interpretation.
| Visualization | Annotation sources | ||||||
|---|---|---|---|---|---|---|---|
| Genome browser | BAM viewer | Population frequency | Structural | Prediction | Evidence based | Local dB | |
| Alamut | | | | | | | |
| Variant Studio | | | | | | | |
| Geneticist Assistant | | | | | | ||
| Ingenuity Variant Analysis | | | | | |||
| Visualization | Annotation sources | ||||||
|---|---|---|---|---|---|---|---|
| Genome browser | BAM viewer | Population frequency | Structural | Prediction | Evidence based | Local dB | |
| Alamut | | | | | | | |
| Variant Studio | | | | | | | |
| Geneticist Assistant | | | | | | ||
| Ingenuity Variant Analysis | | | | | |||
| Visualization | Annotation sources | ||||||
|---|---|---|---|---|---|---|---|
| Genome browser | BAM viewer | Population frequency | Structural | Prediction | Evidence based | Local dB | |
| Alamut | | | | | | | |
| Variant Studio | | | | | | | |
| Geneticist Assistant | | | | | | ||
| Ingenuity Variant Analysis | | | | | |||
| Visualization | Annotation sources | ||||||
|---|---|---|---|---|---|---|---|
| Genome browser | BAM viewer | Population frequency | Structural | Prediction | Evidence based | Local dB | |
| Alamut | | | | | | | |
| Variant Studio | | | | | | | |
| Geneticist Assistant | | | | | | ||
| Ingenuity Variant Analysis | | | | | |||
The combination of annotations used to prioritize variants requires formulation on an application-specific basis, and as with secondary analysis, multiple similar solutions should often be used in combination to account for inaccuracies. In addition to the commercial tools described in Table 4, there are open-source software tools (46, 47) that integrate data across annotation categories (population frequency, structural based, prediction based, evidence based) allowing for rapid tertiary analysis without complex analytical work flow construction. Regardless of the annotation tools used, if implemented correctly, the various sources of annotation enable often overwhelming large variant lists to be filtered or prioritized, resulting in a more manageable number of potentially relevant variants for follow-up, which often includes human visualization of alignments to verify their status as genuine biological variants.
Clinical Guidelines and Requirements
All clinical bioinformatics systems require that primary, secondary, and tertiary analytical components be properly documented and validated. There are several key entities that define these clinical requirements and ensure compliance, including the Centers for Medicare and Medicaid Services and its Clinical Laboratory Improvement Amendments (CLIA), the College of American Pathology (CAP), and in some instances state regulatory bodies such as the New York State Department of Health. These entities have extended the existing molecular diagnostic accreditation guidelines/requirements to address NGS and the bioinformatics software used for NGS data analysis and interpretation, including the CAP Molecular Pathology Checklist (61) and the New York State “Next Generation” Sequencing guidelines (62). Additionally, academic and governmental organizations have published specific recommendations for best practices in NGS and bioinformatics analysis, including the CDC (38), the American College of Molecular Genetics (39), and the CLSI (63). Together, these requirements and best-practice recommendations provide a framework upon which a clinical bioinformatics pipeline should be constructed to ensure accurate and safe patient testing.
Fundamental to the accreditation requirements and best practice guidelines is extensive documentation of the bioinformatics work flows, processes, validation studies, and QC measures used in the clinical data analysis. Specific requirements include standard operating procedures (SOPs) providing step-by-step instruction for executing the clinical data analysis. The documentation should provide a description of the overall work flow and must include a catalog of all software and version numbers used in the analysis process. Additionally, this must include a clear description of all nondefault software configurations and, for reference-based read alignment, documentation of the version and source of origin for all reference sequence data. The CAP checklist and New York State requirements also mandate that all QC and variant calling parameters are documented and justified and routine monitoring data captured. Finally, if any automated filtering or prioritization processes are used to identify putatively casual variants, these must also be clearly documented. Together these documents should enable a domain expert to enter a clinical sequencing laboratory and understand and execute the data analysis process for each clinical assay, thereby ensuring standardization, reproducibility, and transparency of the clinical test.
Accreditation requirements also state that bioinformatics pipelines must be validated and performance criteria clearly defined. This should include determination of variant calling sensitivity, specificity, accuracy, and precision for all variant types reported by the clinical assay. These metrics are dependent on the minimum read coverage for the reportable range and should be characterized at this level. New York State specifically requires laboratories to define the minimum coverage necessary to call a base position variant or normal at a defined frequency, confidence level, and estimated error rate. It is also important to determine the detection limit of variant length (in bases) for insertions and deletions; this is heavily influenced by sequencing chemistry (read length) and read-mapping algorithms. Assays reporting heterogeneous genotypes (e.g., mitochondrial heteroplasmy and somatic tumor mutations) must determine the limit of variant frequency detection in the assay system for each variant type and for indels, which should be established at the defined length detection threshold. Additionally, target regions with high homology to off-target genomic locations must be identified and tested for lower variant calling accuracies.
To establish the variant calling performance metrics, synthetic data sets with clearly established variant profiles may be used to estimate analytical performance, but ultimately testing should also include well-characterized biological samples. These often will consist of internal laboratory samples characterized by an independent gold-standard technology, such as Sanger sequencing, or widely available public samples, such as those included in the International HapMap project (64) and available through the nonprofit Coriell Institute for Medical Research. Furthermore, NIST created the Genome-in-a-Bottle project to provide a reference sample(s) with high confidence variant calls established across multiple sites, sequencing instruments, aligners, and variant callers (65), thereby enabling uniform assessment of performance between testing laboratories.
There are several additional recommendations and requirements that address the overall bioinformatics and IT processes used in a clinical NGS laboratory. For assays using multiplexed NGS runs, there must be documentation of a clear ability to demultiplex and clearly subset sample-specific reads. There must be defined procedures for identifying software updates, determining whether to implement these updates, and subsequently validating the bioinformatics following the updates. Laboratories may also be required to define data retention policies, describing which files will be retained, for what duration of time, and when archiving processes will take place. The laboratory must maintain a clear exception log, recording all deviations from the SOPs and QC acceptance criteria. In addition, there are accreditation requirements by New York State mandating confirmation of all novel and clinically actionable variants with an independent technology or process due to the nascent nature of NGS in clinical testing and the need to minimize the return of false results. These requirements state that once a distinct target area has a variant type confirmed 10 times, independent confirmation is no longer required. Refinement of this requirement may occur as NGS testing experiences increase and more studies (66) emerge, more clearly defining what mutation types require confirmation and what types may not.
In addition to the accreditation requirements reviewed in this section, there are many additional compliance challenges that can impact bioinformatics. These include, but are not limited to, adherence to HIPAA (Health Insurance Portability and Accountability Act) (67) regulations and protection of patient health information; a challenge when institutions consider alternative computational infrastructures, such as cloud computing. Compliance with the litany of requirements addressing NGS necessitates increased documentation and diligence in the bioinformatics work flows used to analyze patient data. This level of rigor may be foreign to many bioinformatics groups who have traditionally operated in a research-only environment, requiring substantial education and review. In addition, due to the emerging nature of NGS in the clinical laboratories these policies and best practice recommendations are in a continued state of flux, making adherence nontrivial. Despite these challenges, strict following of accreditation requirements is necessary to maintain a safe and compliant clinical laboratory.
Challenges and Future Directions
As described, NGS technologies have progressed from a research tool to a diverse clinical platform (68) in a relatively short time span. Such success has only been possible due to an appreciation of the technology's shortcomings and compensation for these in implementing clinical assays. Short read lengths, high error rates, time-consuming or expensive protocols, and bioinformatics deficiencies have all been addressed to varying degrees, enabling successful clinical deployment of the technology. Nonetheless, diverse challenges still exist and represent obstacles to expanded and improved levels of clinical utility.
Characterization of larger genomic aberrations represents a current gray area in genomic profiling. The term “indel” is often used to classify insertions or deletions under 50 bases in length, with larger events being dubbed either structural or copy number variants. This somewhat arbitrary distinction represents an area of uncertainty in variant calling, in which performance metrics are less confidently defined, partially due to a lack of gold standard data. Although a variety of software solutions exist to detect such variations, they are not generally regarded to be as mature as solutions for smaller variations. Tools generally have poor concordance, and no combination of algorithms is considered adequate to exhaustively profile an individual's structural and copy number variations (36). Improved gold standards enabling better performance characterization of tools aimed at structural and copy number variant analyses are required to enable higher confidence in their clinical deployment. Increased read lengths will also address these challenges in the long term.
Haplotype phasing presents another difficulty in clinical sequencing. Genotype information is typically unphased, meaning that information about a variant's chromosome of origin is not captured. Such knowledge can be important for a variety of reasons, including the detection of compound heterozygous events. Traditional methods of phasing possess limited clinical applicability due to lack of resolution, labor intensiveness, or expense. NGS has the potential to address phasing with various algorithmic approaches under development (69). Despite some success, phasing is still a challenge, with read coverage a major factor in its success rate.
Major histocompatibility complex (HLA) and CYP2D6 sequencing represent major aspirations of clinical NGS efforts. Variation in the HLA gene has relevance in organ transplantation, autoimmune disease, cancer, AIDS, and beyond. CYP2D6 is a member of the cytochrome p450 family and is responsible for the metabolism of over 25% of drugs, with variations in its sequence bestowing varying degrees of drug sensitivity and toxicity upon affected individuals. The genomic regions containing both HLA and CYP2D6 are not only highly polymorphic but pose a challenge due to the presence of repetitive sequences, segmental duplications, deletions, and recombinations (70, 71). The characteristics of these regions makes their accurate clinical profiling difficult, and although progress is being made in the area (71), bioinformatics improvements and increased read lengths are likely necessary to enable NGS to routinely exploit these areas clinically.
The recent release of human reference genome GRCh38 by the Genome Reference Consortium represents an alternative form of challenge. The new reference is the first genome release in over 4 years and incorporates many changes from the previous version, including increased representation of pericentromeric regions, alternate sequence representation for variable regions, and the correction of several thousand bases believed to be errors or minor alleles in the previous sequence. Such extensive differences extend to the many resources that annotate the genomic sequence and form essential components of any genomics-based work flow. Dependent on application, reannotation of legacy results to ensure compatibility with the new genome release may be necessary, creating a work burden on clinical laboratories. Equally, previously analyzed patients with undiagnosed conditions may require reanalysis against the new genome to determine if sequence or annotation changes result in changes in read mapping and variant calls.
Perhaps the greatest challenge to clinical sequencing efforts is the improved ability to determine the functional relevance of detected variants. The expanding volume of genomic sequence data generates steadily increasing numbers of variants of unknown significance (VUS), particularly in exome- or genome-wide familial studies. These variants have major clinical relevance because they represent potential drivers of disease or targets of treatment, and their significance extends beyond the affected patient when family members or future offspring might be affected by their inheritance. Factors such as cosegregation, population frequency, and functional analysis have been used in the past to better characterize such variation (72), but such data are often sparse, and the large numbers of VUS that are generated are not conducive to low-throughput methods of functional characterization. Noncoding variants are particularly challenging because the majority of bioinformatics methods to date have concentrated on the coding portion of the genome, effectively ignoring >99% of variation. In this area, newer aggregative methods are beginning to look beyond coding sequences while integrating vital knowledge from existing predictive tools and major ongoing initiatives like ENCODE (51, 73). Increasingly, phenotypic information is being exploited to enable improved prioritization of variants on the basis of predicted functional relevance. Such approaches use similarities between an individual's phenotype and data contained within disease and phenotype ontologies to infer the likelihood of a gene's involvement in contributing to an observed trait. Initial efforts in this area have demonstrated substantial improvements in the ability to prioritize causal variants (74, 75). Beyond this, clinical annotation initiatives like ClinVar are in their early stages and will also aid in the evidence-based phenotypic characterization of such variation and the dissemination of resulting knowledge. Continued development along these parallel lines will be vital in expanding the catalog of variants with predictable functional consequence and thus pushing the boundaries of clinical sequencing efforts beyond current limitations and into a new era of applicability and ubiquity.
Conclusions
NGS assays will continue to push the boundaries of genetics and transform clinical testing for the near future. Incumbent on the success of NGS are the bioinformatics algorithms and tools to transform data into actionable knowledge. Current test offerings are advancing from small gene panels to complete genomes, and with these advances comes an increasing need for improved bioinformatics, including analytics, annotations, software to deliver this information, and systems to capture the realized knowledge. The bioinformatician has now become an essential part of genetic testing laboratories, and their transition from research laboratories into the clinical environment is necessary to provide testing frameworks of the highest quality.
2 Nonstandard abbreviations
- NGS
next generation sequencing
- NHLBI
National Heart, Lung, and Blood Institute
- CAP
College of American Pathology
- SOP
standard operating procedure
- VUS
variants of unknown significance.
3 Human genes
- BRCA1
breast cancer 1, early onset
- AKT1
v-akt murine thymoma viral oncogene homolog 1
- CYP2C19
cytochrome P450, family 2, subfamily C, polypeptide 19
- CYP2D6
cytochrome P450, family 2, subfamily D, polypeptide 6
- HLA
major histocompatibility complex.
Author Contributions:All authors confirmed they have contributed to the intellectual content of this paper and have met the following 3 requirements: (a) significant contributions to the conception and design, acquisition of data, or analysis and interpretation of data; (b) drafting or revising the article for intellectual content; and (c) final approval of the published article.
Authors' Disclosures or Potential Conflicts of Interest:Upon manuscript submission, all authors completed the author disclosure form. Disclosures and/or potential conflicts of interest:
Employment or Leadership: None declared.
Consultant or Advisory Role: E.W. Klee, Soft Genetics.
Stock Ownership: None declared.
Honoraria: None declared.
Research Funding: None declared.
Expert Testimony: None declared.
Patents: None declared.
References

{kind=link}
{kind=link}